Authors: M.Ye, H.-S.Kim, J.-W.Kim, C.-J.Won, K.Haule, D.Vanderbilt, S.-W.Cheong and G. Blumberg
Abstract:
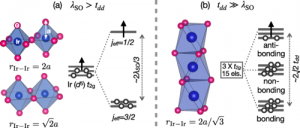
We report ab-initio density functional theory calculation and Raman scattering results to explore the electronic structure of Ba5CuIr3O12 single crystals. This insulating iridate, consisting of face-sharing IrO6 octahedra forming quasi-one-dimensional chains, cannot be described by the local jeff = 1/2 moment picture commonly adopted for discussing electronic and magnetic properties of iridate compounds with IrO6 octahedra. The shorter Ir-Ir distance in the face-sharing geometry, compared to corner- or edge-sharing structures, leads to strong covalency between neighboring Ir. Then this strong covalency results in the formation of molecular orbitals (MO) at each Ir trimers as the low-energy electronic degree of freedom. The theoretically predicted three-peak structure in the joint density of states, a distinct indication of deviation from the jeff = 1/2 picture, is verified by observing the three-peak structure in the electronic excitation spectrum by Raman scattering.